
Horizontal gene transfer (HGT) and gene duplication are often considered as separate mechanisms driving the evolution of new functions. However, the mobile genetic elements (MGEs) implicated in HGT can copy themselves, so positive selection on MGEs could drive gene duplications. Here, we use a combination of modeling and experimental evolution to examine this hypothesis and use long-read genome sequences of tens of thousands of bacterial isolates to examine its generality in nature. Modeling and experiments show that antibiotic selection can drive the evolution of duplicated antibiotic resistance genes (ARGs) through MGE transposition. A key implication is that duplicated ARGs should be enriched in environments associated with antibiotic use. To test this, we examined the distribution of duplicated ARGs in 18,938 complete bacterial genomes with ecological metadata. Duplicated ARGs are highly enriched in bacteria isolated from humans and livestock. Duplicated ARGs are further enriched in an independent set of 321 antibiotic-resistant clinical isolates. Our findings indicate that duplicated genes often encode functions undergoing positive selection and horizontal gene transfer in microbial communities.
Selection for higher gene expression can promote the rapid evolution of duplicated genes through diverse molecular mechanisms 1,2,3,4,5 . Furthermore, gene duplication has long been recognized as a crucial step in the evolution of new functions and traits 1,6,7 . For these reasons, gene duplication is an important evolutionary mechanism for rapid adaptation to novel metabolic and ecological niches 8,9,10,11,12 . Recently duplicated and thus functionally redundant genes often revert to a single-copy state in the absence of selection 13 , suggesting that selection is required to maintain duplicated genes. Indeed, selection for strong gene expression is a key factor for the preservation of duplicated antibiotic resistance genes (ARGs) on plasmids 14 . In addition, recent metagenomic studies indicate that copy number variation in the human microbiome is common and influences human health 15,16 .
Laboratory experiments have demonstrated that positive selection can drive the rapid evolution of gene duplications, due to the rapid kinetics of molecular mechanisms like tandem amplifications 4,17 . While several studies have examined tandem duplications and gene amplifications under laboratory selection for drug resistance 3,18,19,20 or specific metabolic functions 8,9,11 , few studies have examined the role of mobile genetic elements (MGEs) in promoting gene duplications.
Following Partridge et al. 21 , we define MGEs as “elements that promote intracellular DNA mobility (e.g., from the chromosome to a plasmid or between plasmids) as well as those that enable intercellular DNA mobility”. In our experiments, we focus on transposons and plasmids, which are known to mediate the horizontal transfer of ARGs in microbial communities 5,22 . Our bioinformatics analyses more broadly examine genes encoding MGE components, including genes involved in transposon, integrase, bacteriophage, and plasmid functions.
Previously, we showed that antibiotics select for the movement of transposable ARGs from chromosomes onto multicopy plasmids, because the increased copy number of ARGs on multicopy plasmids leads to higher expression of those genes and thus higher resistance 5 . Based on those findings, we reasoned that antibiotic selection would also favor duplications of ARGs, generated by intrachromosomal transposition events. We tested this hypothesis using mathematical modeling, experimental evolution, and genome sequencing to confirm the location and copy number of transposable ARGs in evolved populations.
Based on these experimental findings, we reasoned that antibiotic use should enrich specific populations of bacteria with duplicated ARGs. Several recent studies have reported cases of gene duplications in clinical antibiotic-resistant isolates, using long-read sequencing or qPCR to measure resistance gene copy number 23,24,25,26,27,28,29,30,31,32,33 . However, it is not known whether these cases represent a broader trend. To address this question, we examined the distribution of duplicated genes in tens of thousands of complete bacterial genomes that were sequenced with long-read sequencing technologies.
To date, few studies have systematically examined duplicated genes in bacterial genomes 34 , due to the difficulty of resolving identical sequence repeats with second-generation short-read sequencing technologies 35 . Such sequence repeats facilitate gene duplication 2 , but also hamper their discovery by short-read sequencing, due to read alignment inaccuracies 36 . These issues also plague genome assembly from complex metagenomic samples 37 . Long-read sequencing is critical because long reads can span repeat regions, including transposons and duplicated genes. This resolves ambiguities in copy number variation, including the coexistence of plasmids, in a given isolate or metagenomic sample 35,38 .
Here, by combining modeling, experiments, and bioinformatic analyses, we show that MGEs serve as potent drivers of gene duplications, that gene duplications mediated by MGEs are often adaptive, that duplicated ARGs are enriched in isolates from humans and livestock (the microbial environments most associated with antibiotic use), that duplicated ARGs are further enriched in clinical antibiotic-resistant isolates, and that duplicated ARGs are far more likely to be associated with MGEs than single-copy ARGs. These findings indicate that duplicated genes often encode functions undergoing positive selection and horizontal gene transfer in microbial communities.
Our basic intuition is that mutants with a duplicated ARG can invade an ancestral clonal population with a single-copy resistance gene, given a sufficiently high concentration of antibiotic. To formalize this idea, we built a mathematical model (Fig. 1A, Supplementary Data 1) based on the framework in our previous study 5 . This model involves three subpopulations of bacteria: the first carries an ARG on the chromosome (Type 1), the second has a duplicated ARG on the chromosome (Type 2), and the third carries a duplicated ARG on a plasmid (Type 3). The ARG confers a fitness benefit in the presence of antibiotics due to resistance, and additional copies confer stronger resistance. However, the additional copies may incur a fitness cost in the absence of antibiotic. We assume that all cells contain a plasmid. By letting the copy number of the plasmid be a free parameter of the model, we can also model the no plasmid case (plasmid copy number = 0). The fitness of each population therefore depends on antibiotic concentration, the cost of ARG expression, and the effective number of ARG copies per cell in each subpopulation, which depends on plasmid copy number (Methods: Mathematical model: Fitness functions).
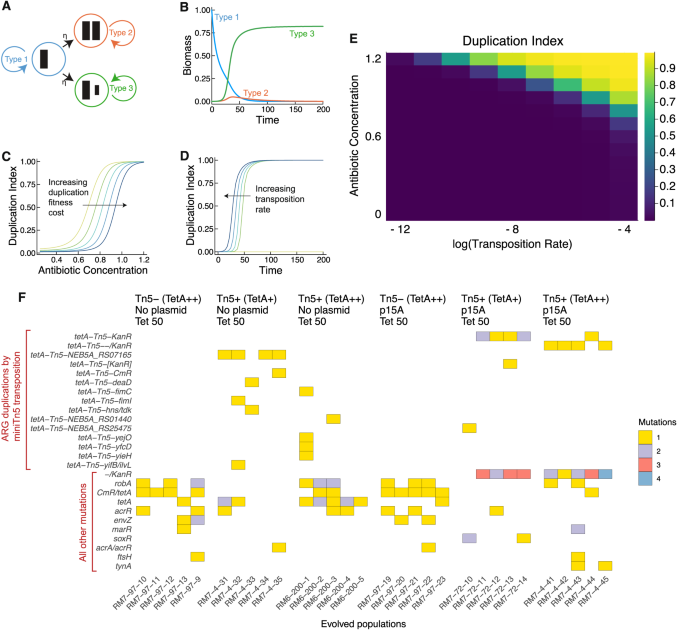
Under antibiotic selection, one of the subpopulations with the additional ARG copy rapidly outcompetes the others, depending on which has the highest fitness. When the cost of expressing additional ARG copies is low, then the Type 3 subpopulation, which contains duplicated ARGs on the plasmid, dominates (Fig. 1B). When the cost of expressing the ARG on the plasmid outweighs the benefit of resistance, the Type 2 subpopulation, which contains duplicated ARGs on the chromosome, dominates (Supplementary Data 1). By defining a “Duplication Index” as the fraction of the population with a duplicated ARG, we find that duplicated ARGs rapidly establish throughout the population at a threshold antibiotic concentration. As the cost of ARG expression increases, this threshold concentration increases. This is shown by the rightward shift of curves representing higher ARG expression costs in Fig. 1C. In addition, as the transposition rate of the transposable ARG increases, the time for establishment of duplicated ARGs in the population decreases, as shown by a leftward shift of curves representing higher transposition rates in Fig. 1D. Furthermore, the model shows that for any given ARG expression cost, duplicated ARGs will establish in the population when both the transposition rate and antibiotic concentration are sufficiently high (Fig. 1E). Altogether, these results highlight what the dynamics of antibiotic selection and ARG duplication could look like, and illustrate a basic model that can be tested experimentally.
We tested the core prediction of this model— that antibiotics select for duplicated ARGs— by carrying out evolution experiments with E. coli strains harboring a minimal transposon composed of a tetA tetracycline resistance gene flanked by 19-base-pair terminal repeats. This mini-transposon is mobilized by an external Tn5 transposase in the chromosome 39 . We carried out 9-day selection experiments with E. coli DH5α and sequenced populations resistant to 50 μg/mL tetracycline, varying plasmid, the presence of active transposase, and the basal expression of the tetA resistance gene. We also evolved and sequenced a parallel set of control populations that were propagated without tetracycline (Supplementary Data 2). Multiple transpositions of the tetA-Tn5 transposon to both chromosome and plasmid are observed in the presence of active transposase. In the absence of active transposase, we see parallel mutations affecting the tetA promoter and the native efflux pump regulatory genes robA, marR and acrR (Fig. 1F). By contrast, no gene duplications were observed in the no-antibiotic control populations, nor was any parallel evolution observed (Supplementary Data 2). This finding implies that tetracycline treatment selected for the tetA duplications and the other resistance mutations observed across replicate populations (Fig. 1F).
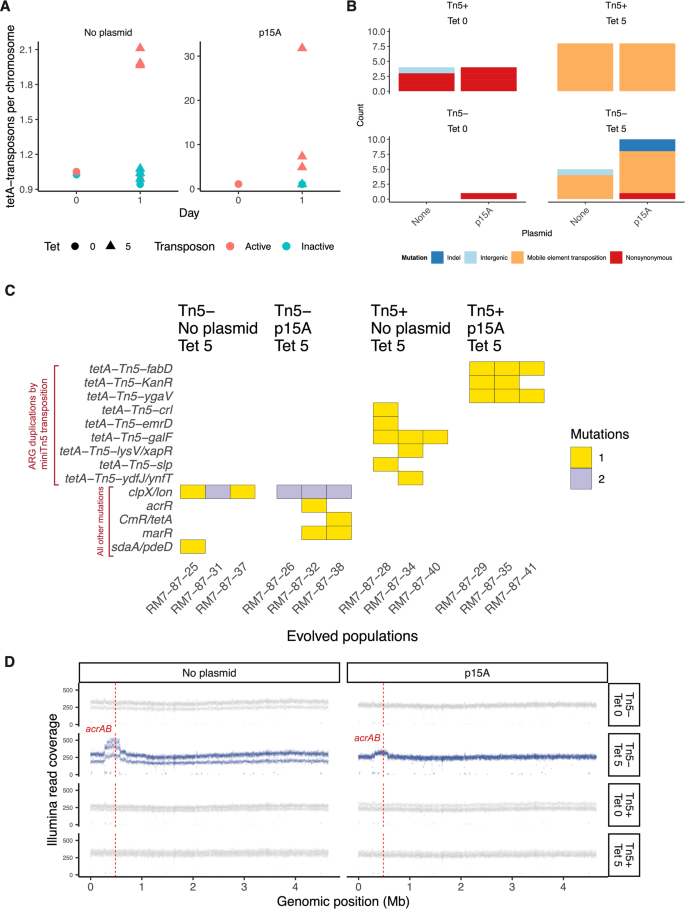
Given this finding, we asked whether duplications could arise as a short-term evolutionary response, in a wild-type K-12 MG1655 genetic background. Given the high activity of the synthetic tetA-Tn5 transposon, one day of tetracycline selection ( ~ 10 bacterial generations) was sufficient to drive duplications of the tetracycline resistance gene to observable allele frequencies across all replicate populations, both in the presence and absence of plasmids (Fig. 2A). By contrast, no duplications were observed in the no-antibiotic control populations (Figure 2A, B, C, D). No tetA duplications were observed in the absence of transposase, although gene amplifications of the native acrAB antibiotic efflux pump were seen (Fig. 2D). Since no tetA duplications or other resistance mutations were observed in the no-antibiotic control treatment (Supplementary Data 2), we infer that tetracycline treatment directly selected for the observed tetA duplications, acrAB amplifications, and other resistance mutations. We then replaced the tetA gene in the minimal Tn5 transposon with smR, kanR, ampR, and cmR genes conferring resistance to spectinomycin, kanamycin, carbenicillin, and chloramphenicol, and repeated our one-day selection experiment using these four antibiotics. ARG duplications were observed in 8 out of 8 evolved populations, across all four antibiotic treatments (Supplementary Fig. 1). Together, the mathematical model and these evolution experiments demonstrate the that antibiotic selection can drive the evolution of duplicated ARGs via intragenomic transposition.
To examine the relevance of duplicated ARGs in the ecological context of natural and clinical isolates, we downloaded all complete and fully annotated bacterial genomes from NCBI RefSeq 40 passing additional quality control checks (25,224 genomes were downloaded and 24,102 genome passed quality control, see Methods: Curation of complete bacterial genomes) and grouped them into 7 different ecological categories (excluding “Unannotated”) based on their isolation source and host source metadata (Supplementary Data 3). We used categories similar to, but with higher granularity than, the ProGenomes2 Database 41 . We then examined the distribution of duplicated ARGs across these 7 ecological categories, spanning 18,938 genomes after excluding those that were assigned to the “Unannotated” category (Supplementary Data 3). We define “duplicated” genes based on 100% amino-acid sequence identity. Therefore, our analysis calls a pair of genes within a genome that only differ by silent (synonymous) substitutions “duplicated”, while a pair of genes that differ by a single amino-acid change would be called as a pair of “single-copy” genes (Fig. 3).
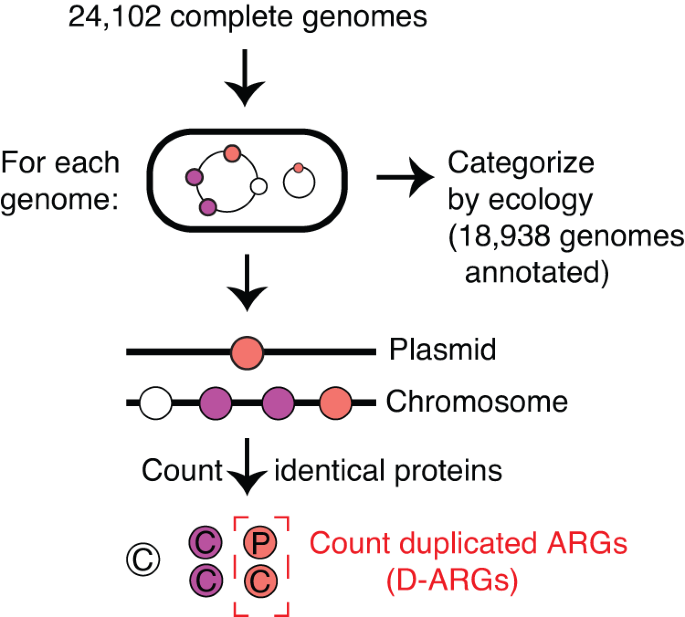
The 100% sequence identity threshold is critical for defining duplicated genes. When a protein is encoded by two separate loci in the genome, one can assume that its production is redundant. This assumption is much harder to justify if the two copies differ by even a small number of amino acid substitutions, since those may nevertheless have substantial effects on protein function. Given the redundant production of a protein at two or more loci, one can suppose one of two possibilities. Either the duplication event has occurred in the recent past, such that not enough time has passed for the two copies to diverge in sequence, or the production of the protein from multiple loci may be evolving under strong purifying selection, such that the sequence found at multiple loci is being preserved as time passes.
Our operational definition of duplicated genes does not take plasmid copy number into account, such that a protein encoded on a multi-copy plasmid would be classified as “single-copy” if there is no additional sequence encoding the same protein elsewhere in the genome. While modifying our definition such that all plasmid-borne proteins count as “duplicated” does not change our conclusions, it has the disadvantage of collapsing the useful distinction between proteins encoded once or multiple times on a plasmid.
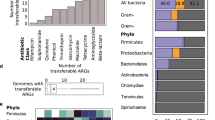
We estimated the proportion of isolates carrying duplicated ARGs in each ecological category: this estimate represents the empirical probability of whether an isolate from a given ecological category has duplicated ARGs. Isolates from humans and livestock show significantly higher proportions of isolates carrying duplicated ARGs, in comparison to the other categories (Fig. 4A and Supplementary Table 1). This trend holds for many different classes of antibiotics, including chloramphenicol, tetracycline, MLS antibiotics, beta-lactams, diaminopyrimidines, sulfonamides, quinolones, aminoglycosides, and macrolides. (Supplementary Fig. 2). By comparison, most isolates in all categories have at least one annotated ARG (Fig. 4B, Supplementary Table 2), and at least one duplicated gene (Fig. 4C and Supplementary Table 3). This result holds for duplicated ARGs found solely on chromosomes or plasmids (Supplementary Fig. 3).
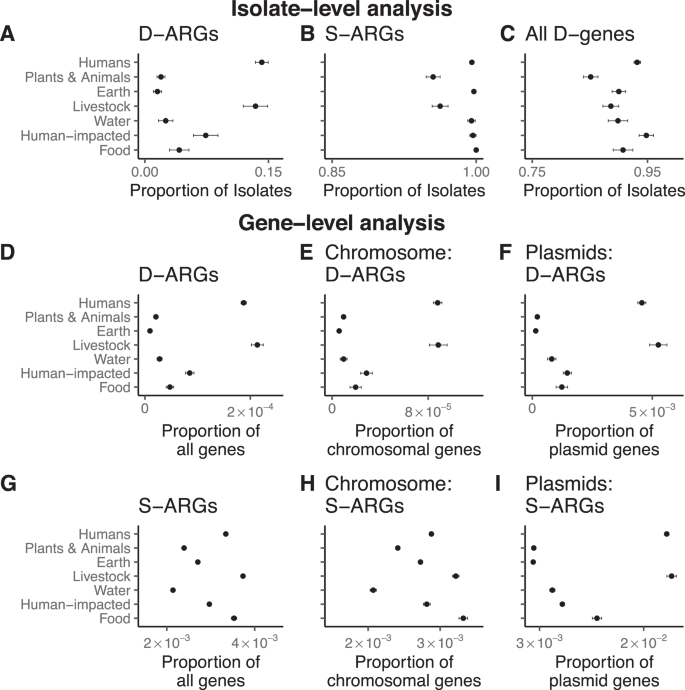
We checked the robustness of the pattern shown in Fig. 4A with further computational controls. We reasoned that the association between duplicated ARGs and isolates from humans and livestock could be affected by both the over-representation of some bacterial taxa, as well as phylogenetic correlations between highly related isolates. To evaluate these possibilities, we compared the number of isolates per bacterial genus to the number of isolates containing duplicated ARGs per bacterial genus. Klebsiella and Escherichia are over-represented among both the isolates as well as isolates containing duplicated ARGs. Several other genera containing human commensals and pathogens (Staphylococcus, Salmonella, Pseudomonas, Acinetobacter) are highly represented and often have duplicated ARGs (Supplementary Fig. 4A). After removing the bacterial genera that are most enriched with isolates containing duplicated ARGs, the overall difference between categories is much smaller, although isolates from livestock are still most likely to contain duplicated ARGs (Supplementary Fig. 4B). Within the genera that are most enriched with isolates containing duplicated ARGs, isolates from humans and livestock are still much more likely to contain duplicated ARGs (Supplementary Fig. 4C). To examine the effect of phylogenetic correlations between highly related isolates, we downsampled the data in two ways. First, we used Assembly Dereplicator 42 to remove genomes based on a pairwise phylogenetic distance threshold (Mash distance > 0.005). Second, we downsampled the data to one genome per species. After downsampling, isolates from humans and livestock are still most likely to contain duplicated ARGs compared to the other categories (Supplementary Fig. 4D, E). This analysis indicates that the association between duplicated ARGs and isolates from humans and livestock is robust, but most relevant for a small number of bacterial genera. Within those genera, strains isolated from humans and livestock are much more likely to carry duplicated ARGs.
We also examined all the genes, rather than the isolates, in each ecological category. Although genes within a genome have correlated evolutionary histories due to vertical descent, this analysis provides additional context for our main results, and uses a methodology that is consistent with metagenomic studies that focus on the abundance of genes and their functional annotations, rather than genomes per se, as ecological markers 43 . Duplicated ARGs encompass a much higher proportion of genes in the human-host and livestock categories in comparison to the other ecological categories (Fig. 4D, Supplementary Table 4). This trend holds for both chromosomal genes (Fig. 4E) as well as for plasmid genes (Fig. 4F). The gene-level analysis also shows that single-copy ARGs are frequent in the human-host and livestock categories (Fig. 4G, Supplementary Table 5), again for both chromosomal genes (Fig. 4H) and plasmid genes (Fig. 4I). When examining separate classes of antibiotics, we find that single-copy tetracycline and sulfonamide resistance genes are most common in the human-host and the livestock category (Supplementary Fig. 5).
To validate the medical relevance of these findings, we searched the recent literature for datasets of bacterial genomes satisfying three criteria: (1) High quality, publicly available, and fully annotated genomes sequenced by long-read technologies; (2) known provenance from clinical antibiotic-resistant isolates; and (3) independence from our main dataset of complete genomes from NCBI RefSeq, to rigorously test the hypothesis that antibiotic treatment selects for duplicated ARGs. We found four genomic datasets satisfying these criteria and measured the extent to which each dataset contained duplicated ARGs. First, we re-examined the genomes of 12 clinical extended-spectrum beta-lactam (ESBL) resistant E. coli isolates from Duke University Hospital, that were previously sequenced by our group and colleagues 44 . 6 of these 12 isolates contain duplicated ARGs (Supplementary Fig. 6). Second, we also examined the genomes of 46 ESBL-resistant and vancomycin-resistant Enterobacter, Escherichia, and Klebsiella that were sequenced as part of the BARNARDS study of antibiotic resistance at 12 clinical sites in 7 countries across Africa and South Asia 45 . 23 of these 46 isolates contain duplicated ARGs (Supplementary Fig. 7). Third, we examined the genomes of 149 clinical ESBL-like E. coli isolates from a tertiary care hospital 46 . 36 of these 149 isolates contain duplicated ARGs (Supplementary Fig. 8). Fourth, we examined the genomes of 114 clinical ESBL-resistant isolates from an Australian ICU 47 . 20 of these 114 isolates contain duplicated ARGs (Supplementary Fig. 9). Altogether, 26% of these clinical antibiotic-resistant isolates (85 out of 321) contain duplicated ARGs. By contrast, 14% of the human isolates in our main dataset (1054 out of 7490) contain duplicated ARGs (Fig. 2A and Supplementary Table 1). Therefore, the clinical antibiotic-resistant isolates in these additional datasets are enriched with duplicated ARGs, relative to the general human isolates in our main dataset (Binomial test: p < 10 −8 ).
The clinical genomes isolated from an Australian ICU (NCBI BioProject PRJNA646837) had complete and fully annotated plasmid sequences, so we examined plasmid copy number relative to chromosome across this set of clinical ESBL-resistant strains 47 . Plasmids carrying beta-lactamases had significantly higher copy number than plasmids carrying other kinds of resistance genes (Mann-Whitney U-test, p < 10 –16 ). However, plasmids carrying ARGs had significantly lower copy numbers than plasmids without ARGs (Mann-Whitney U-test, p < 10 –16 ). Regardless, these data show that plasmid copy number tends to increase the copy number of linked ARGs (Supplementary Fig. 10).
If ARGs evolve additional copies under selection for increased gene dosage, then we expect that ARGs, especially those associated with MGEs, would often occur on plasmids, because plasmids often have a higher copy number than the chromosome. We tested this prediction by comparing the distribution of single-copy ARGs on chromosomes and plasmids to the distribution of duplicated ARGs on chromosomes and plasmids (Fig. 4E, F, H, I).
Single-copy ARGs also show strong associations with plasmids (Supplementary Table 5). 189,137 single-copy ARGs occur on chromosomes, while 23,315 occur on plasmids, in comparison to 67,078,963 non-ARG single-copy genes on chromosomes and 1,967,705 non-ARG single-copy genes on plasmids. In this case as well, we find an overwhelming association between single-copy ARGs and plasmids (Fisher’s exact test: p < 10 –16 ). Therefore, the statistical association between ARGs and plasmids is general. These results also show that in terms of absolute numbers, most single-copy ARGs occur on chromosomes, while most duplicated ARGs occur on plasmids.
When we examine the functional annotation of duplicated genes (Methods: Sequence classification based on functional annotation), we find that ~60% (608,465 out of 977,928 duplicated genes) are associated with MGE components, such as genes involved in transposon, integrase, bacteriophage, and plasmid functions (Fig. 5A). This finding is intuitive, since this class of genes often encode components of “DNA cut-and-paste” and “DNA copy-and-paste” machinery. This trend holds for both duplicated genes found on chromosomes (539,878 out of 853,702 duplicated chromosomal genes) as well as for those found on plasmids (68,587 out of 124,226 duplicated plasmid genes), and this trend holds across all ecological categories. By contrast, less than 5% of single-copy genes on chromosomes encode functions related to MGEs (2,511,319 out of 67,268,100 single-copy chromosomal genes), while ~15% of single-copy genes on plasmids encode MGE-related functions (316,013 out of 1,991,020 single-copy plasmid genes) (Fig. 5B).
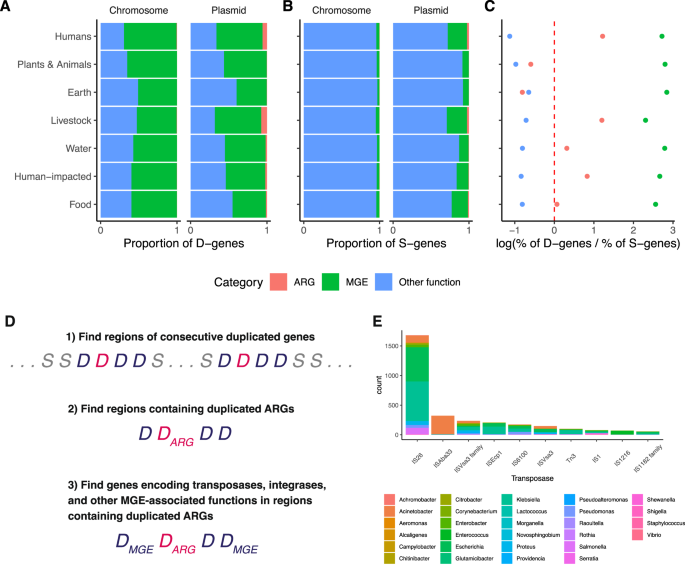
Suppose no evolutionary forces such as selection, horizontal gene transfer, or associations with MGEs affect the probability that a gene undergoes gene duplication. Under this null hypothesis, the probability that a gene of a given functional class is duplicated should be proportional to the fraction of single-copy genes represented by this functional class. Deviations from this null expectation (i.e., the ratio of the proportion of duplicated genes to the proportion of single-copy genes equals one, implying that the log-ratio equals zero) indicates that the frequency of duplicated genes is being driven away from equilibrium by evolutionary forces. A visual explanation of this method and the null expectation is shown in Supplementary Fig. 12.
Using this method, we find that duplicated ARGs are enriched in bacteria isolated from humans, livestock, water, and human-impacted environments; fit the null expectation for bacteria isolated from food, and are depleted from plants, animals, and earth (Fig. 5C). Duplicated MGE-associated genes are highly enriched across all environments. Furthermore, duplicated genes encoding all other functions are depleted across all environments (Fig. 5C). This test indicates that duplicated ARGs are being driven to higher-than-expected frequencies in bacteria isolated from humans, livestock, water, and human-impacted environments, due to some evolutionary force like selection, horizontal gene transfer, or both.
To investigate linkage between duplicated ARGs and genes encoding MGE functions, we conducted two analyses. First, we asked whether duplicated ARGs had a higher probability of being flanked by MGE-associated genes, in comparison to single-copy ARGs. This was indeed the case. Examining ARGs across all 18,938 genomes, we found that 4651 out of 9836 duplicated ARGs were flanked by MGE-associated genes, while 37,181 out of 278,074 single-copy ARGs were flanked by MGE-associated genes. Therefore, duplicated ARGs are far more likely than single-copy ARGs to be linked with MGE-associated genes (Binomial test: p < 10 –16 ).
Second, we examined regions of consecutive duplicate genes in each of the 18,938 genomes (Fig. 5D). 3356 regions contain duplicated ARGs and duplicated MGE-associated genes, while 2551 regions contain duplicated ARGs but no duplicated MGE-associated genes. Therefore, annotated MGE-associated genes, such as transposases, are an important factor but are not required for ARG duplication. Of these 6087 regions, 237 contain multiple copies of some duplicated ARG. Therefore, segmental duplications account for a relatively small fraction of duplicated regions in these data. We also compared the relative frequency of transposases and phage integrases in the duplicated regions containing ARGs. 8449 genes encode MGE functions within the duplicated regions containing ARGs. Of these, 5541 encoded transposases. By comparison, 1046 encoded integrases. Therefore, transposases make up a large fraction of the duplicated MGE-function genes associated with duplicated ARGs. Among these, the IS26 transposase has particular significance 48 (Fig. 5E and Supplementary Fig. 13). IS26 is known to play a major role in the spread of diverse ARGs, including associations with antibiotic resistance plasmids found in carbapenem-resistant Klebsiella pneumoniae 49,50,51 .
Our modeling demonstrates that ARG duplication could be an effective mechanism for the evolution of antibiotic resistance. Our genomic analyses show that MGEs, such as the transposons and plasmids in our experiments, can serve as a vehicle for the duplication of ARGs. This finding has relevance for natural and clinical populations, as demonstrated by our bioinformatic analyses. Specifically, the distribution of duplicated ARGs in bacterial genomes isolated from different environments is shaped by non-random evolutionary forces, such as antibiotic selection. This evolutionary process is likely facilitated by association with MGEs. Together, these results imply that antibiotic usage not only enriches for resistant subpopulations: it also selects for mutants with a higher capacity for evolutionary innovation through gene duplication, because one gene copy can maintain ancestral function, while additional copies are free to evolve new functions 1,6 . Our results indicate that MGEs have an intrinsic ability to drive evolutionary innovation through their ability to catalyze the duplication and HGT of passenger genes, such as ARGs, carried within the MGE 52,53 .
This work implies that gene duplication in bacteria is often linked to horizontal gene transfer, through a common dependence on MGEs. This conclusion contrasts with previous studies that have treated gene duplication and horizontal gene transfer as distinct mechanisms for genetic innovation in bacteria 54,55 . Our work also contrasts with the majority of experimental studies on gene duplications in bacteria 3,4,14,18,19,56 , which have focused on tandem amplifications—and not MGE transposition— as a driver of gene duplication in bacteria 5,57 . A key limitation of our study, however, is that we do not directly identify MGEs in our bioinformatic analysis, due to the technical challenge of doing so comprehensively, reliably, and rapidly across all complete bacterial genomes. The development of databases and tools to identify MGEs across the tree of life will allow researchers to measure the extent to which MGEs contribute to the duplication, diversification, and horizontal transfer of genes under positive selection.
The enrichment we observe of duplicated ARGs in humans and livestock is most likely caused by high rates of antimicrobial exposure 58 . Indeed, our analysis of clinical antibiotic-resistant strains strongly supports antibiotic use as a primary driver for the evolution of duplicated ARGs— even though we do not know the resistance phenotypes or antibiotic treatment history for most of the genomes in this study. Future research could examine the quantitative relationship between antibiotic use and the evolution of duplicated ARGs in settings such as hospitals 59 and factory farms 60,61 .
Our analysis has several caveats that warrant analysis in future research. First, our mathematical model implicitly assumes that the mutation rate for genomic resistance mutations is small compared to ARG transposition rates— small enough that genomic resistance mutations can be ignored. More work is needed to measure how the relative magnitudes of these rates, and the relative selective benefits of these molecular mechanisms, affects the evolution of antibiotic resistance by ARG duplication. Second, our experiments focused on E. coli, and did not examine whether MGEs promote gene duplication across bacterial species. Given our bioinformatics results, we expect our experimental findings to hold across bacteria, but direct experimental tests are needed. For instance, we expect that the transposition rates may depend on idiosyncratic interactions between a given MGE and its host. In this case, it would be interesting to ask whether the prevalence of a given MGE in a particular bacterial species can predict the transposition rate of that MGE in that species, and thus the importance of particular MGEs for spreading clinically relevant resistance in different pathogen species. Third, while we examined several different ARGs in our experiments, it is unclear whether the type of resistance mechanism encoded by an ARG (e.g., antibiotic degradation, target modification, efflux pump activity) affects the likelihood of resistance evolution by gene duplication. Given the generality of our bioinformatic results across multiple classes of antibiotics (Supplementary Fig. 2), we predict that the specific molecular mechanism of resistance has little impact on ARG duplication dynamics. Indeed, our mathematical model predicts that the dynamics of duplicated ARGs only depends on transposition rates and the balance of fitness benefits and costs of expressing duplicated ARGs, which needs to be tested by future experiments with a broader set of ARGs operating with different mechanisms.
Finally, our results suggest that duplicated genes, especially those encoded on plasmids, may represent a signature of ongoing horizontal gene transfer and adaptation in microbial communities. If so, one could identify genes under ongoing HGT and natural selection by quantifying gene duplication. For instance, it would be both interesting and important to test whether microbial communities in the permafrost of the Arctic tundra show novel genomic patterns of copy number variation in response to climate change 62 , and to test whether pathogens and their hosts show characteristic patterns of copy number variation as they coevolve 63 . Our results also suggest that researchers can compress a bacterial genome into a set of dozens of duplicated genes, while maintaining important evolutionary and ecological information about ongoing HGT and selection. Such simple and practical techniques for producing reduced summaries of biological datasets 64 would allow researchers to scale population genomic analyses of microbial communities and their HGT networks 62,65,66,67 to millions of microbial genomes and plasmids.
We built a mathematical model, based on the framework used by Lopatkin et al. 68 and by ref. 5 , to study how antibiotic usage can select for duplicated ARGs. A diagram of the model is shown in Fig. 1A. This model involves three subpopulations of bacteria: the first carries an ARG on the chromosome (Type 1), the second has a duplicated ARG on the chromosome (Type 2), and the third carries a duplicated ARG on a plasmid (Type 3). We are interested in the dynamics of the three subpopulations due to selection (growth and dilution) and mutation (duplication by transposition dynamics of the ARG).
See Supplementary Data 1 for an interactive Pluto computational notebook of the model written in Julia 1.8. This notebook can be run by installing and running Pluto.jl within Julia 1.8+ (see instructions at: https://plutojl.org/) and then opening the notebook using the Pluto web browser interface. Unless otherwise stated, the simulation results shown in Fig. 1 use the following default parameter settings (arbitrary units): Antibiotic Concentration A = 2.0, Duplication Cost c = 0.1, Transposition Rate η = 0.0002, Dilution Rate D = 0.1, Plasmid copy number y = 2.
We assume that there is a steady inflow of nutrients and antibiotic, and a steady outflow of depleted media and cells, reflected by a constant dilution rate, D. This assumption allows the population to grow continuously at a steady-state population size. We normalize the number of cells by the carrying capacity, such that each state variable represents the percentage of carrying capacity that is taken up by the subpopulation– note that this is not the relative frequency of cells in the population, because the total population may be at a steady state that is less than carrying capacity. The growth rate of each subpopulation is modeled by growth functions fi > 0, that we describe in greater detail below.
We define a mutation as a transition from one state to another due to gene duplication by transposition. Each transition occurs at a constant rate η. We assume that transposon excision rates are negligible, such that duplication events leave the original copy unchanged in the chromosome.
These assumptions lead to a system of differential equations of the form:
$$\frac<>_>>=__\left(1-\Sigma _\right)-D_+_$$where the first term reflects logistic growth at rate fi, the second term reflects constant dilution due to a fixed outflow rate, and the third term wraps up all the state transitions (mutation dynamics).